Энергетика и химическая природа катализа
В соответствии с современными физико-химическими представлениями о сущности катализа катализатор и реагирующие вещества следует рассматривать как единую каталитическую реакционную систему, в которой химические превращения испытывают не только реактанты под действием катализатора, но и катализатор при взаимодействии с реагентами. В результате такого взаимного воздействия в реакционной системе устанавливается стационарный состав поверхности катализатора, определяющий его каталитическую активность. Отсюда следует, что катализатор — не просто место осуществления реакции, а непосредственный участник химического взаимодействия, и его каталитическая активность обусловливается химической природой катализатора и его химическим сродством к реактантам.
Исходя из основного постулата о химической природе взаимодействия в каталитической реакционной системе можно сформулировать некоторые важные для предвидения каталитического действия термодинамические и кинетические принципы.
- Катализатор должен химически взаимодействовать хотя бы с одним из компонентов реагирующих веществ (с образованием координационных, ионных или ковалентных связей).
- Изменение свободной энергии процессов взаимодействия в каталитической реакционной системе должно быть менее отрицательным, чем изменение свободной энергии катализируемой реакции, т. е. соединения реагирующих веществ с катализатором должны быть термодинамически менее прочными, чем продукты реакции (если это требование не соблюдается, катализатор быстро выходит из строя, образуя нерегенерируемое прочное химическое соединение).
- Многостадийный каталитический процесс термодинамически будет наиболее выгодным (вероятным), если изменения свободной энергии на каждой из стадий примерно одинаковы и равны половине изменения теплового эффекта суммарного процесса.
- В кинетическом отношении каталитическая реакция будет идти с большей скоростью, если в результате промежуточного химического взаимодействия катализатор будет снижать энергию активации химической реакции (или одновременно повышать предэкспонент Аррениуса). Это правило согласуется с принципом компенсации энергии разрывающихся связей в катализе. Оно согласуется также с принципом энергетического соответствия мультиплетной теории А. А. Баландина.
- Установлена определенная закономерность между специфичностью каталитического действия и типом кристаллической структуры твердых тел. Каталитической активностью ионного и электронного типов обладают твердые тела соответственно с ионной и металлической кристаллической структурой, а также кристаллы промежуточного (ионно-металлического) типа. Молекулярные и ковалентные кристаллы в отношении катализа практически инертны.
Ионный катализ. Катализаторами в ионном (гетеролитическом) катализе являются кислоты и основания. Каталитическая активность кислот и оснований обусловливается способностью их к обмену реагирующей молекулой ионом или парой электронов с образованием промежуточного соединения ионного типа, обладающего высокой реакционной способностью.
Согласно протонной теории Бренстеда и Лоури, кислота и основание — вещества (нейтральные молекулы или ионы), являющиеся соответственно донором или акцептором протона, т. е.
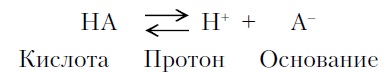
В теории Бренстеда-Лоури отличительным признаком кислоты считается наличие в ее молекуле протона. Эта теория не рассматривает проявления кислотного характера веществами, не содержащими водорода, например SnCl4, BF3, AlCl3, ZnCl2, алюмосиликата, цеолита и др. Недостатки протонной теории устранены и дополнены в электронной теории кислот и оснований Льюиса.
По электронной теории Льюиса кислотой и основанием являются вещества, являющиеся соответственно акцептором и донором электронных пар. Льюисовские кислоты (L-кислоты) и основания могут не содержать протонов и, следовательно, являются апротонными. Кислотно-основное взаимодействие заключается в образовании донорноакцепторной связи типа

Большинство катионов являются L-кислотами, а анионов — льюисовскими основаниями. Соли — типичные кислотно-основные комплексы. Как видно, электронная теория Льюиса рассматривает вопрос
о кислотах и основаниях более широко, чем другие теории.
Наиболее типичным примером реакций, протекающих по механизму общего кислотного катализа, являются каталитические превращения углеводородов нефти, имеющие место в таких важных в нефтепереработке процессах, как каталитический крекинг, изомеризация и алкилирование.
Апротонные кислоты Льюиса (AlCl3, BF3, ZnCl, SbF4) часто катализируют те же реакции, что и протонные кислоты Бренстеда, причем активность апротонных кислот иногда выше, чем протонных. Обусловливается это тем, что в водных средах (например, в каталитическом крекинге в присутствии водяного пара) апротонные кислоты превращаются в протонные:
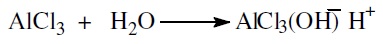
При взаимодействии с кислотами углеводороды ведут себя как слабые основания. Из всех классов углеводородов наибольшей основностью обладают алкены, при этом основность изоалкенов выше.
Полициклические арены являются значительно более сильными основаниями по сравнению с моноциклическими. Алканы характеризуются наименее слабой основностью.
В нефтепереработке принято называть образующиеся при взаимодействии углеводородов с кислотным катализатором первичное (промежуточное) соединение карбений-ионом или карбкатионом, а катализ — соответственно карбений-ионным.
Карбкатионы наиболее легко образуются при передаче протона от бренстедского кислотного катализатора к молекуле олефина, который может образоваться при термолизе углеводородов:
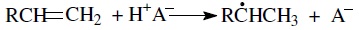
Надо отметить, что протон Н+ (гидрид-ион, гидрид-радикал Н) характеризуется исключительно высокой реакционной способностью, что объясняется отсутствием у него электронной оболочки. Гидрид-ион —
единственный катион, не имеющий электрона. Диаметр Н+ примерно в 104 раз меньше диаметра любого другого катиона.
Карбкатион, образующийся при взаимодействии протона с олефином, называют карбений-ионом. Термин «карбоний-ион», часто неправильно используемый в литературе, относят к карбкатиону, образующемуся в результате присоединения протона к парафину:
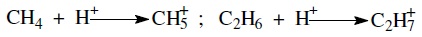 и т.д.
и т.д.
При атаке протоном олефина π-электроны двойной связи используются для образования новой σ-связи между протоном и одним из углеродных атомов, образующим двойную связь, при этом второй углеродный атом углеводорода заряжается положительно. Таким образом, карбений-ион является промежуточной структурой между олефином, имеющим π-связь, и парафином, в котором есть только σ-связь.
При взаимодействии олефина с протоном возможно образование двух разных карбений-ионов:
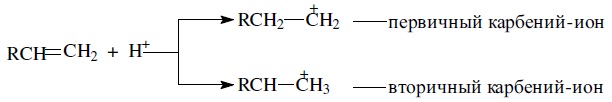
Расчеты показывают, что теплота образования первичных карбениевых ионов на 81 кДж/моль больше, чем для вторичных, и на 81 + 61 кДж/моль больше, чем для третичных. Вследствие этого первичные карбениевые ионы быстро переходят в третичные.
Карбениевые ионы являются высокоактивными частицами, вступающими во вторичные реакции с углеводородами с исключительно большой скоростью. Активность (константа скорости) карбений-ионов
на несколько порядков выше активности радикалов.
Основными реакциями карбкатионов, как и радикалов, являются мономолекулярный распад по Р-правилу и бимолекулярные реакции замещения и присоединения. Существенное отличие карбкатионов от радикалов — способность первых к изомеризации, что объясняется значительным снижением свободной энергии при переходе от первичного к вторичному и третичному карбкатионам.
Электронный катализ. В электронном (окислительно-восстановительном) катализе ускоряющее действие катализаторов достигается облегчением электронных переходов в гемолитических реакциях за счет свободных электронов переходных металлов.
Переходные металлы являются активными катализаторами в подавляющем большинстве окислительно-восстановительных реакций. Железо, например, является классическим катализатором синтеза аммиака. Кобальт, никель, медь и металлы платиновой группы проявляют высокую активность в процессах гидрирования и дегидрирования, а также окисления. Серебро является практически единственным катализатором парциального окисления (например, этилена до его окиси).
Характерной особенностью переходных металлов является незавершенность их электронных d-оболочек, определяющая их специфические химические (переменная валентность, склонность к комплексообразованию), многие физические (образование кристаллов металлического типа, работа выхода электрона из металла, электропроводимость, магнитные свойства и др.) и каталитические свойства.
В кристаллическом состоянии часть электронов из d-оболочек переходит в зону проводимости и возникает возможность обмена электронами между d- и внешней s-оболочкой. Энергетическая легкость подобного перехода (определяемая работой выхода электрона из металла) приводит к тому, что на внешней поверхности кристалла образуется определенное число свободных электронов. Их наличие приводит к появлению на поверхности свободных валентностей — положительных в случае свободного электрона (электронно-донорная проводимость) и отрицательных при отсутствии электрона (электронно-акцепторная, так называемая «дырочная» проводимость) у частицы, расположенной на поверхности кристалла.
Наличие свободных валентностей на поверхности электронных катализаторов определяет прежде всего их адсорбционные (хемосорбционные) свойства. При этом возможны два различных механизма процесса хемосорбции.
- Поверхность катализатора обладает меньшим сродством к электрону адсорбирующегося атома или молекулы, как, например, хемосорбция кислорода на металлической поверхности. В этом случае возникает ковалентная связь за счет перехода свободных электронов из металла к кислороду (т. е. кислород является окислителем).
- Поверхность металла обладает большим сродством к электрону, по сравнению со сродством к электрону адсорбирующегося атома. Типичный пример — хемосорбция водорода на металлической поверхности (например, платины). В этом случае происходит переход электрона от адсорбирующейся молекулы в металл (водород является восстановителем).
Бифункциональный катализ имеет место в других промышленно важных процессах, в которых одни стадии сложной реакции протекают по ионному, а другие — электронному катализу. По такому ионно-электронному катализу осуществляются реакции ароматизации (дегидроциклизации) нормальных алканов и пятичленных нафтенов в процессе каталитического риформинга бензина, реакции деструктивного гидрирования в процессе гидрокрекинга, а также изомеризации С4–С6 алканов.
Естественно, катализаторы бифункционального катализа должны содержать в своем составе одновременно оба типа центров — и металлические (м. ц.), и кислотные (к. ц.). Так, полиметаллический алюмоплатиновый катализатор риформинга представляет собой платину, модифицированную редкоземельными металлами (например, Re), на носителе — окиси алюминия, промотированном кислотой (хлором). В катализаторе гидрокрекинга, например алюмокобальтмолибденцеолитовом (или алюмоникельмолибденцеолитовом), Со + Мо или Ni + Mo осуществляют гидрирующе-дегидрирующие функции, а цеолит является кислотным компонентом. В качестве примера приведем возможные схемы протекания подобных реакций.
- Реакция дегидроциклизации нормального гексана:
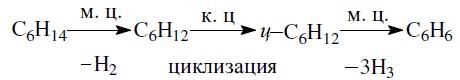
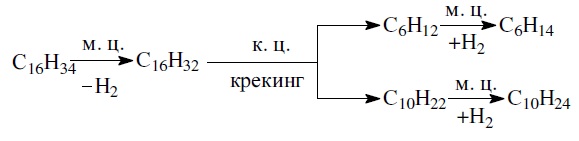
- Реакция гидрокрекинга С16Н34:
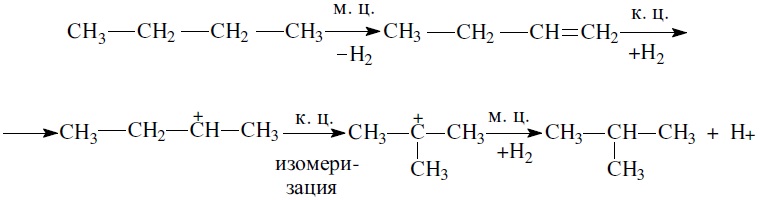
- Реакции изомеризации н-бутана
ТЕХНОЛОГИЯ И ОБОРУДОВАНИЕ ПРОЦЕССОВ ПЕРЕРАБОТКИ НЕФТИ И ГАЗА, С. А. Ахметов, Т. П. Сериков, И. Р. Кузеев, М. И. Баязитов, 2006