Механизм и химизм каталитического крекинга
Из изложенных выше закономерностей катализа и анализа физикохимических свойств катализаторов и сырья крекинга можно констатировать, что:
- химические превращения крекируемого сырья осуществляютсяn по карбений-ионному механизму посредством хемосорбции молекул углеводородов к поверхности катализатора, состоящего из слабоактивной крупнопористой матрицы из алюмосиликата и из активного компонента — цеолита;
- оба участника каталитической реакционной системы характеризуются неоднородностью по реакционной способности: неоднородность поверхности катализатора обусловливается наличием каталитических центров различной силы кислотности, следовательно, активности, а сырье крекинга неоднородно по молекулярной массе и химическому составу;
- каждый акт хемосорбции осуществляется обменом протоном между катализатором и реактантом, причем нет принципиальной разницы между протонами, отщепляемыми из цеолита или из алюмосиликата. Процесс хемосорбции может начаться с отрыва протона на одних центрах и закончиться с возвратом протона на другие центры катализатора. Следовательно, в каталитическом химическом процессе может иметь место миграция хемосорбированных молекул по поверхности катализатора;
- каталитический процесс может осуществляться посредством точечной или мультиплетной (особенно реакции скелетной изомеризации) хемосорбции;
- более вероятно, что за один акт хемосорбции химическая реакция не завершается с образованием конечного продукта: она осуществляется многостадийно, т. е. по цепному механизму, через образование и последующие превращения промежуточных веществ;
- поскольку поверхность цеолитов, имеющих поры малых размеров, недоступна для диффузии крупных молекул исходного сырья, первичные химические реакции, например крекинга или деалкилирования, должны протекать преимущественно на поверхности матрицы катализатора.
Химические превращения углеводородов крекируемого сырья, протекающие по карбений-ионному цепному механизму на поверхности ЦСК, можно представить в целом в следующей последовательности.
- Первичные мономолекулярные реакции крекинга и деалкилирования (распад по С–С-связи) высокомолекулярных молекул исходного сырья с образованием низкомолекулярных (н. м.) углеводородов:
а) крекинг парафинов с образованием н. м. парафина и олефина:
СnH2n+2 → CmH2m + CpH2p+2 ;
б) крекинг олефинов с образованием н. м. олефинов:
СnH2n → CmH2m + CpH2p ;
в) деалкилирование алкилароматических углеводородов:
ArCnH2n+1 → ArH + CnH2n → ArCmH2m–1 + CpH2p ;
г) крекинг нафтенов с образованием олефинов:
ц-СnН2n → CmH2m + CpH2p ,
где n = m + р.
Первичные реакции распада могут осуществляться либо термически по радикально-цепному механизму, либо каталитически на апротонных (льюисовских) центрах алюмосиликатной матрицы ЦСК:
RH + L → R+ + LH–
R+ → н. м. олефин + R+'
R+' + LH → R'H + L или
R+' → H+ + олефин
- Вторичные бимолекулярные реакции углеводородов на поверхности цеолита с участием карбений-ионов, образующихся преимущественно присоединением протона к олефину (инициирование цепи):
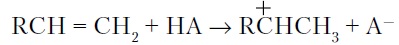
Различие по реакционной способности образующихся карбкатионов обусловливает вероятные направления превращений и степень участия их в дальнейших реакциях. Установлено, что стабильность карбениевых ионов возрастает в ряду:
СН3 < +С2Н5 < + первичный < вторичный < третичный.
Третичный карбениевый ион является самым стабильным. Именно этим обусловлен высокий выход изопарафиновых углеводородов, особенно изобутана, при каталитическом крекинге.
Реакции развития цепи включают следующие наиболее характерные реакции карбениевых ионов: распад С–С-связи, перенос гидридиона (Н-перенос), изомеризацию, циклизацию, дециклизацию, деалкилирование, алкилирование, полимеризацию, поликонденсацию и др.
Обрыв цепи превращений карбениевых ионов происходит возвратом протона к поверхности катализатора или отнятием электрона от центров Льюиса.
Распад С–С-связи карбений-иона является одной из наиболее важных целевых реакций, приводящих к образованию низкомолекулярных топливных фракций и С3–С4 углеводородов в газах каталитического крекинга. Для этой реакции применимы следующие правила:
- легче всего разрывается С–С-связь, находящаяся в β-положении по отношению к атому углерода, несущему заряд (правило — β-распада);
- у образующихся олефинов имеется двойная связь у первого углеродного атома;
- из нескольких возможных вариантов более вероятен β-распад карбений-иона с образованием олефина с меньшей длиной цепи:
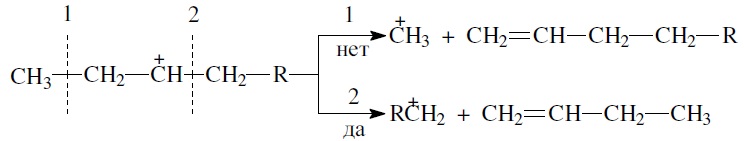
Продукт первичного β -распада — карбений-ион RC+H2 — может снова крекироваться до образования более стабильных карбкатионов или углеводородов (после отдачи протона или присоединения электрона);
- более выгодным для алкилароматических или алкилнафтеновых углеводородов является отрыв всей алкильной группы:
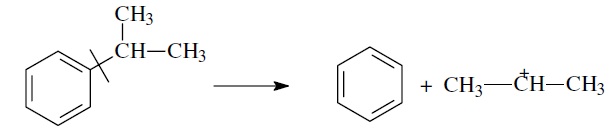
Поскольку образование С+H3 и С+2H5 требует высоких энергетических затрат, цепной распад карбкатионов прерывается до образования карбениевых ионов с числом углеродных атомов 3…5.
Перенос гидрид-иона (Н-перенос) можно проиллюстрировать следующим образом:
R1H + R+ → R+1 + R2H
Установлено, что лучшими гидридными донорами являются нафтены, полициклические нафтены или нафтено-ароматические углеводороды, изоалканы и даже олефины. Энергетически более выгоден отрыв гидрид-иона от третичного, затем вторичного и менее выгоден от первичного углеродного атома. Нафтеновые, алкилароматические и изопарафиновые углеводороды часто содержат третичные атомы углерода и поэтому интенсивно участвуют в реакциях Н-переноса. Активными акцепторами гидрид-ионов являются наименее стабильные высокореакционноспособные карбений-ионы или углеводороды, содержащие несколько π-связей, например диолефины. Именно Н-перенос обусловливает повышенные выход топливных фракций и химическую стабильность бензинов каталитического крекинга. По Н-переносу осуществляются следующие реакции каталитического крекинга:
олефин + нафтен → парафин + арен
олефин + парафин → парафин + олефин
олефин + олефин → арен + парафин
олефин + олефин → арен + водород
арен + арен → кокс + парафин + водород и т. д.
Изомеризация карбениевых ионов является наряду с распадом важной целевой реакцией, повышающей товарные качества продуктов каталитического крекинга.
В большинстве случаев изомеризация протекает быстрее, чем крекинг, и потому часто предшествует β-распаду. Сочетание реакций изомеризации и β-распада обусловливает повышенное содержание в продуктах каталитического крекинга углеводородов изостроения.
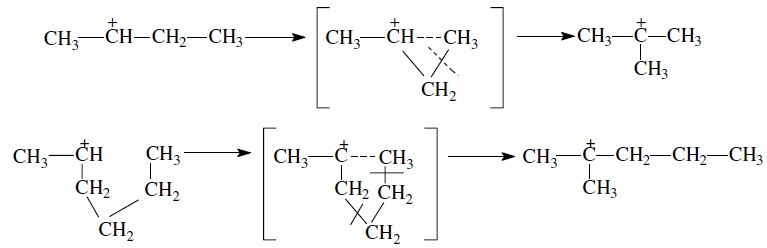
Изомеризация карбениевых ионов может происходить либо путем передачи протона (гидридный сдвиг), либо метильной группы (скелетная изомеризация) вдоль углеводородной цепи:

Для реакций изомеризации предложен механизм, согласно которому процесс осуществляется через образование промежуточных циклических структур, например циклопропана, циклобутана и т. д. (по-видимому, посредством многоточечной, т. е. мультиплетной хемосорбции):
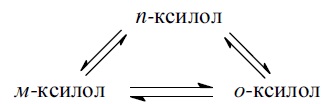
и переносом метильной группы внутри молекулы при изомеризации ди- и полиметилбензолов. Так, ксилолы подвергаются взаимопревращению:
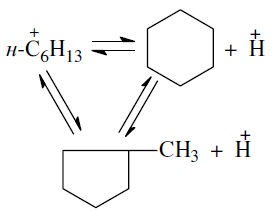
Циклизация и дециклизация как обратимые реакции с участием карбений-ионов протекают, по-видимому, через мультиплетную хемосорбцию:
или через диеновый синтез:
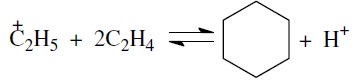
Циклопентаны в условиях каталитического крекинга более устойчивы, чем циклогексаны. Циклогексаны в этих условиях могут подвергаться дегидрированию в арены посредством Н-переноса.
При наличии длинных боковых цепей в циклоалкановом карбениевом ионе возможны изомеризация боковой цепи и деалкилирование.
Бициклические циклоалкановые карбениевые ионы ароматизируются в большей степени, чем моноциклические.
Алкилирование и полимеризация — реакции, противоположные крекингу, протекают по карбений-ионному механизму. При температурах ниже 400 °С они доминируют над крекингом, а при высоких температурах равновесие смещается в сторону деалкилирования и деполимеризации.
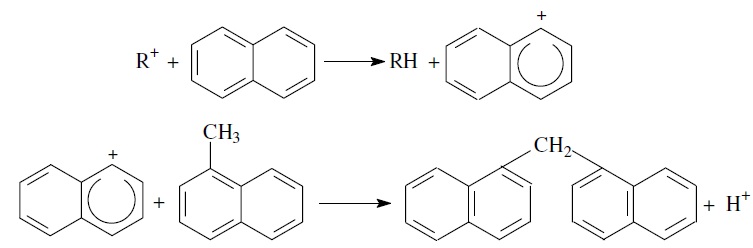
Конденсация ароматических углеводородов, дающая соединения с более высокой молекулярной массой, вплоть до кокса, характерна для каталитического крекинга. При этом ареновый карбений-ион вступает в последовательные реакции присоединения (конденсации) к ароматическим углеводородам и Н-переноса. Процесс конденсации вследствие высокой стабильности полициклического аренового карбений-иона может продолжаться до обрыва цепи:
Коксообразование. При осуществлении реакций углеводородов на кислотных катализаторах образуется углеродистый материал, называемый коксом, который не десорбируется с поверхности катализатора. Этот материал имеет атомное отношение водорода к углероду от 0,3 до 1,0 и спектроскопические характеристики, аналогичные таковым для полициклических ароматических соединений.
При крекинге ароматических углеводородов кокс получается более обогащенным углеродом, чем при крекинге парафинистого сырья. В составе кокса крекинга сернистого нефтяного сырья всегда содержится сера. В среднем отношение содержания серы в коксе к ее содержанию в сырье крекинга близко к единице.
Вследствие экранизации активных центров ЦСК коксовыми отложениями активность катализатора крекинга быстро снижается. Эта дезактивация является обратимой, так как после окислительной регенерации первоначальная активность практически полностью восстанавливается. При этом тепло регенерации полезно используется для обеспечения теплового баланса в системе. Кроме того, образующийся при выводе из сырья избытка углерода водород полезен в реакциях Н-переноса, тем самым для увеличения выхода бензина на сырье и повышения его химической стабильности.
Образующийся при крекинге нефтяного сырья кокс принято подразделять на четыре типа:
- «каталитический» кокс, который образуется на кислотных катализаторах;
- «дегидрогенизационный» кокс, образующийся в результате реакций дегидрирования на металлах, осадившихся из сырья;
- «хемосорбционный» кокс, получающийся в результате необратимой хемосорбции высококипящих полициклических аренов и смолистоасфальтеновых компонентов сырья (т. е. связанный непосредственно с коксуемостью сырья);
- «десорбируемый» кокс, остающийся в порах катализатора в результате неполной десорбции в отпарных зонах реакционных аппаратов.
Ниже приведен примерный выход на катализаторе каждого из типов в общей массе образующегося кокса, % отн.
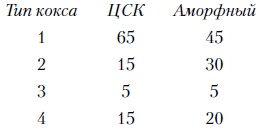
Образование «каталитического» кокса непосредственно связано с реакциями циклизации олефинов, конденсации, алкилирования и Н-переноса. Полициклические арены, олефины и полиолефины более коксогенны, чем парафины и нафтены.
Коксообразующая способность полициклических аренов возрастает при увеличении их числа в ряду бензол → нафталин → антрацен и в ряду бензол → дифенил → терфенил.
Интенсивность образования «дегидрогенизационного» кокса определяется содержанием и типом отлагающегося на катализаторе металла сырья. Наибольший выход этого типа кокса обеспечивают кобальт, никель, медь и в меньшей степени ванадий, молибден, хром и железо. Интенсивность образования кокса, помимо свойств катализатора и химического состава сырья, определяется также кинетическими параметрами технологического процесса.
Из сопоставления качества продуктов можно указать на следующие преимущества каталитического крекинга на ЦСК перед термическим:
- каталитический процесс протекает более селективно и приводит к преимущественному образованию С3–С4 углеводородов в газах, в то время как в газах термического крекинга преобладают С1–C2 углеводороды;
- благодаря более интенсивному протеканию реакций изомеризации (двойных связей и скелетной) и ароматизации в продуктах каталитического крекинга содержится значительно больше алканов и алкеновизостроения и ароматических углеводородов;
- в продуктах каталитического крекинга благодаря реакциям Н-переноса отсутствуют диолефины и содержится значительно меньше моноолефинов;
- каталитический процесс позволяет получить бензины с более высокими октановым числом и химической стабильностью и большим выходом.
ТЕХНОЛОГИЯ И ОБОРУДОВАНИЕ ПРОЦЕССОВ ПЕРЕРАБОТКИ НЕФТИ И ГАЗА, С. А. Ахметов, Т. П. Сериков, И. Р. Кузеев, М. И. Баязитов, 2006